
Obsah článku
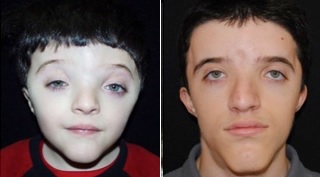 Saethre-Chotzenův syndrom patří do skupiny vzácných genetických poruch známých jako poruchy „akrocefalosyndaktylie“.
Saethre-Chotzenův syndrom patří do skupiny vzácných genetických poruch známých jako poruchy „akrocefalosyndaktylie“.
Akrocefalosyndaktylie jsou skupinou vrozených vývojových vad s akrocefalií a syndaktylií.
Všechny tyto nemoci jsou charakterizovány předčasným uzavřením lebečních švů (to jsou švy mezi určitými kostmi lebky), čemuž se říká kraniosynostóza a / nebo srůstem (či fúzí) určitých prstů na rukou nebo nohou (tomu se říká syndaktylie). U mnoha kojenců se Saethre-Chotzenovým syndromem se mohou lebeční švy spojit nerovnoměrně, což může přispět k tomu, že se hlava a obličej budou z různých stran zdát odlišné (kraniofaciální asymetrie).
Mohou být přítomny i další problémy lebky a obličeje (kraniofaciální), například široce rozmístěné oči (oční hypertelorismus) s neobvykle mělkými očními dutinami (orbity); pokles horních víček (ptóza); a stav, kdy oči nesměřují stejným směrem (strabismus). Někteří postižení jedinci mohou mít také jakoby „zobákový“ nos; problémy s přepážkou, která odděluje nosní dírky; malé, nízko posazené uši; a nedostatečně vyvinutou horní čelist (hypoplastická čelist).
Porucha je také spojena s problémy rukou a nohou, jako je částečná fúze (tj. srůst) měkkých tkání (kožní syndaktylie) určitých prstů na rukou a nohou; neobvykle krátké prsty (brachydaktylie); a široké prsty na nohou. Inteligence je obvykle normální.
Saethre-Chotzenův syndrom se dědí autozomálně dominantním způsobem.
Saethre-Chotzenův syndrom a jeho příznaky
Saethre-Chotzenův syndrom je primárně charakterizován předčasným uzavřením lebečních švů mezi určitými kostmi v lebce (tomu se říká kraniosynostóza), výraznými problémy v oblasti obličeje a / nebo problémy u prstů na rukou a nohou. Související příznaky a nálezy se však mohou velmi lišit, a to i u postižených členů stejné rodiny. Existují například rodiny, ve kterých někteří členové rodiny měli charakteristické abnormality prstů, zatímco jiní byli primárně postiženi kraniosynostózou. Je to hodně individuální.
Pokud je kraniosynostóza přítomna, stupeň malformace lebky (lebeční) se může měnit, v závislosti na konkrétních zúčastněných kraniálních švech a pořadí a rychlosti progrese. U mnoha postižených kojenců a dětí předčasné uzavření koronálního švu, který se nachází mezi kostmi tvořícími čelo (čelní kost) a horními stranami lebky, způsobí, že horní část hlavy vypadá špičatě (akrocefálně) nebo hlava vypadá neobvykle krátce nebo široce (brachycephaly).
Kromě toho lebeční švy často srůstávají nerovnoměrně, což způsobuje, že hlava a obličej vypadají poněkud odlišně z jedné a druhé strany (plagiocefalie a asymetrie obličeje). Byly také hlášeny případy, kdy hlava vykazuje trojúhelníkový tvar (trigonocefalie) nebo čelo je neobvykle výrazné kvůli předčasnému uzavření švu přední kosti (tj. přední neboli metopické švy – metopický šev je umístěn ve středu čela a sahá od velké fontanely ke kořeni nosu). V některých případech může předčasné uzavření určitých lebečních švů vést k abnormálně zvýšenému tlaku v lebce (intrakraniální tlak).
Mnoho jedinců se Saethre-Chotzenovým syndromem má další kraniofaciální variace, jejichž výsledkem je jemný, ale výrazný vzhled obličeje. Takové abnormality mohou zahrnovat široké čelo s nízkou linií vlasů; pokles horních víček (ptóza); zobákový nos, odchýlená nosní přepážka; neobvykle široké, ploché oblasti na obličeji; a malá horní čelist (hypoplastická čelist). Často jsou také přítomny další oční abnormality, jako jsou široce rozmístěné oči (oční hypertelorismus); mělké oční dutiny (oběžné dráhy); strabismus; a / nebo abnormální zúžení slzných cest (stenóza slzného kanálu), potenciálně způsobující snížené slzení a zvýšenou náchylnost k očním infekcím.
S poruchou mohou také souviset další kraniofaciální problémy (kraniofaciální = týkající se lebky a obličeje). Mnoho postižených jedinců má malé, nízko posazené uši. Kromě toho je častá mírná porucha sluchu. Abnormality v oblasti úst (orální) často zahrnují vysoce klenutou střechu úst (patra) a vady zubů, jako je absence nebo malformace určitých zubů, přítomnost nadbytečných zubů a / nebo nesprávný kontakt zubů. Ve vzácných případech může dojít k rozštěpu patra.
Sponzorováno
Saethre-Chotzenův syndrom může být také charakterizován různými problémy prstů na rukou a nohou. Někteří postižení jedinci mají částečný srůst neboli fúzi měkkých tkání (kožní syndaktylie) určitých prstů, zejména mezi druhým a třetím prstem. Méně často se však syndaktylie rozprostírá od druhého po čtvrtý prst nebo zasahuje jiné prsty. Mezi další malformace prstů mohou patřit neobvykle krátké prsty na rukou a nohou (brachydaktylie); a / nebo široké prsty na nohou.
Se Saethre-Chotzenovým syndromem mohou také souviset další fyzické abnormality. Někteří postižení jedinci mají třeba nižší vzrůst. Méně často mohou být také přítomny muskuloskeletální abnormality, jako je srůst neboli fúze určitých kostí páteře v oblasti krku (krční obratle), abnormální fúze kostí předloktí (radioulnární synostóza), omezené prodloužení loktů nebo kolen, krátké klíční kosti a / nebo deformity kyčle (coxa valga). Příležitostné další nálezy mohou zahrnovat neschopnost varlat sestoupit do šourku (kryptorchismus) u postižených mužů; abnormality ledvin; a / nebo srdeční vady.
Většina jedinců se Saethre-Chotzenovým syndromem má normální inteligenci. Někdy je však přítomno mírné až střední mentální postižení.
Příčiny Saethre-Chotzenova syndromu
U většiny jedinců je Saethre-Chotzenův syndrom způsoben mutacemi genu TWIST1.
Saethre-Chotzenův syndrom je autozomálně dominantní stav. Dominantní genetické poruchy se vyskytují, když k vyvolání určité nemoci stačí pouze jedna kopie abnormálního genu. Abnormální gen může být zděděn od jednoho z rodičů nebo může být výsledkem nové mutace (změna genu) u postiženého jedince. Riziko přenosu abnormálního genu z postiženého rodiče na potomstvo je 50% (1 z 2) pro každé těhotenství. Riziko je stejné pro muže i ženy.
U některých jedinců je porucha způsobena spontánní (de novo) genetickou mutací, která se vyskytuje ve vajíčku nebo spermatu. V takových situacích není porucha zděděna po rodičích.
Diagnóza Saethre-Chotzenova syndromu
Diagnóza Saethre-Chotzenova syndromu je primárně založena na fyzických známkách a příznacích. Molekulárně genetické testování mutací v genu TWIST1 lze identifikovat u některých, ale ne u všech jedinců.
Saethre-Chotzenův syndrom se vyskytuje u 1 z 25 000 až 1 z 50 000 novorozenců. Mírná forma poruchy se někdy nazývá Robinow-Soraufův syndrom.
Léčba Saethre-Chotzenova syndromu
Léčba Saethre-Chotzenova syndromu zaměřena na identifikaci potenciálně lékařsky důležitých projevů u jednotlivce a na léčbu těchto projevů. Taková léčba může vyžadovat koordinované úsilí týmu lékařů, jako jsou pediatři; chirurgové; lékaři, kteří diagnostikují a léčí poruchy kostry, kloubů, svalů a příbuzných tkání (ortopedové); lékaři se specializací na poruchy uší, nosu a krku (otolaryngologové); lékaři, kteří diagnostikují a léčí neurologické poruchy (neurologové); a / nebo jiní zdravotničtí pracovníci.
V prvním roce života lze doporučit chirurgický zákrok, který má pomoci předcházet nebo korigovat předčasné uzavření kraniálních švů, má zabránit zvýšenému nitrolebnímu tlaku a zabránit progresivní asymetrii obličeje. Může být také provedena korekční a rekonstrukční chirurgie, která pomůže napravit určité kraniofaciální malformace a související nálezy, syndaktylii, jiné kostní vady nebo jiné fyzické abnormality potenciálně spojené s poruchou. Prováděné chirurgické zákroky budou záviset na závažnosti a umístění anatomických abnormalit, jejich přidružených symptomech a dalších faktorech.
Doporučuje se i vyšetření očních a zrakových abnormalit oftalmologem a audiologické hodnocení ztráty sluchu. RTG krčních obratlů by mělo být zváženo přibližně ve věku 2 let. V závislosti na nálezech jednotlivce mohou být zváženy další testy.
Pokud je identifikováno mentální postižení, může být důležitá včasná intervence, aby se zajistilo, že děti s Saethre-Chotzenovým syndromem dosáhnou svého potenciálu. Mezi speciální služby, které mohou být prospěšné, patří speciální vzdělávání a / nebo jiné lékařské, sociální nebo odborné služby.
Sponzorováno
Genetické poradenství bude také přínosem pro postižené jedince a jejich rodiny.
Studie o tomto syndromu a zdroje článku
Sponzorováno
Autorem článku je naše redakce
Líbil se vám náš článek? Sdílejte ho, uděláte nám radost
Štítky: Genetická onemocnění, Vzácná onemocnění
Přečtěte si také naše další články